Applications/CytoskeletonAnalyzer2D: Difference between revisions
(→Usage) |
|||
| (23 intermediate revisions by the same user not shown) | |||
| Line 3: | Line 3: | ||
== Cytoskeleton Analyzer 2D == | == Cytoskeleton Analyzer 2D == | ||
[[File:MiToBo_logo_CytoskeletonAnalyzer2D.png|border|150px|left|link=]] | |||
[[File:ActinExample.png|150px|right|link=]] | [[File:ActinExample.png|150px|right|link=]] | ||
[[File:HT144-shC-Series010-clusters.png|150px|right|link=]] | [[File:HT144-shC-Series010-clusters.png|150px|right|link=]] | ||
| Line 34: | Line 36: | ||
* run MiToBo and start the operator runner by selecting the menu item '''''MiToBo Runner''''' from '''''Plugins -> MiToBo''''' | * run MiToBo and start the operator runner by selecting the menu item '''''MiToBo Runner''''' from '''''Plugins -> MiToBo''''' | ||
* in the selection menu navigate to 'de.unihalle.informatik.MiToBo.apps.cytoskeleton' and select the operator '''''CytoskeletonAnalyzer2D''''' | * in the selection menu navigate to 'de.unihalle.informatik.MiToBo.apps.cytoskeleton' and select the operator '''''CytoskeletonAnalyzer2D''''' | ||
This will bring up the operator window of the CytoskeletonAnalyzer2D. | This will bring up the operator window of the CytoskeletonAnalyzer2D.<br><br> | ||
* Input data:<br>The operator expects a special organization of the input image data. All the data should be contained in common top-level folder.<br> The images for each treatment/genotype/protein have to be put into | * '''<u>Input data:</u>'''<br>The operator expects a special organization of the input image data which is shown in the figure below. All the data should be contained in a common top-level folder, here named ''"experiment"''.<br> The images for each treatment/genotype/protein have to be put into separate sub-folders of this top-level folder, here named ''"group-1"'', ''"group-2"'' and so on. Besides the set of corresponding images,<br> each sub-folder is required to contain an additional sub-folder named ''"results_segmentation"'' where the annotation files with the cell areas or boundaries, respectively, are stored.<br><br> | ||
* Output data:<br>The operator | [[File:FolderStructure.png|1200px|center|link=]] | ||
:For each image named ''"<imagename>.tif"'' a corresponding mask file is expected to be found in that folder. The mask file should have the same basename like the corresponding image file,<br> but either end on "-mask.tif" (e.g., ''"<imagename>-mask.tif"'') in case of using label images as masks, or on ''"-mask.zip"'' or ''"-mask.roi"'' (e.g., ''"<imagename>-mask.zip"''), respectively, in case of using region sets as mask data.<br> Note that the operator currently only accepts '''ImageJ 1.x ROI sets''' as input. If label images are used as segmentation masks, the labels of individual cells in the label images are used as unique identifiers for the cells, i.e.,<br> are used as labels in the various plots. If the segmentation data is given in terms of ROIs a unique identifier is derived from the order of the cell boundaries in the ROI set.<br> Note that in the latter case a mask image is written to the output directory as additional output where the cells are marked by their identifiers to allow for easier assessment of the results.<br><br> Each of the sub-folders in the top-level folder is treated as a separate group of cells or experimental condition, thus, the cells of all images within one sub-folder are collected in a single set.<br> The names of the folders are used to label the different groups in the various output data files.<br> If the input images contain more than one channel you can select the channel on which the Cytoskeleton Analyzer should work. By default the first channel is used. <br><br> | |||
* '''<u>Output data:</u>'''<br>The operator generates image- or group-specific output data files as listed in the table below. All data files specific to a single input image are stored in a new sub-folder named ''"results_features"'' in the sub-folder of each group. All global output data files are stored directly in the top-level folder. The string ''imageID'' below represents the name of a single input image, the string ''groupID'' refers to a specific group. In addition to saving the output data to file bar charts and box-whisker plots are directly shown in the graphical user interface of ImageJ/Fiji upon termination of the analysis process. <br> <br> | |||
{|class="wikitable" style="margin: auto;" | |||
|style="color:black; background-color:#ffffcc; width:25%"|'''Output File Name''' | |||
|style="color:black; background-color:#ffffcc; width:20%"|'''Where to find it...''' | |||
|style="color:black; background-color:#ffffcc; width:45%"|'''Description''' | |||
{|class="wikitable" | |||
|Name | |||
|Description | |||
|- | |- | ||
|'' | |<imageID>-features.txt | ||
| | |folder ''results_features'' | ||
|Feature data for single image file. | |||
|- | |- | ||
|'' | |<imageID>-features.tif | ||
| | |folder ''results_features'' | ||
|Image stack visualizing the feature data for single image. | |||
|- | |- | ||
|'' | |<imageID>-clusterDistro.txt | ||
| | |folder ''results_features'' | ||
|Cluster distributions for each cell individually and for all cells in total of the image. | |||
|- | |- | ||
|'' | |<imageID>-clusters.tif | ||
| | |folder ''results_features'' | ||
|Pseudo-colored image illustrating the cluster distribution per image. | |||
|- | |- | ||
|'' | |<groupID>-distributionChart.png | ||
| | |folder ''results_features'' | ||
|Stacked bar plot of the cluster distribution for each cell of the group. | |||
|- | |||
|AllCellsClusterStats.txt | |||
|top-level folder | |||
|Cluster distribution of raw data for all images and cells. | |||
|- | |||
|AllCellsPCASubspaceStats.txt | |||
|top-level folder | |||
|If PCA is applied to the cluster distribution vectors this file contains the subspace feature vectors for all cells. | |||
|- | |||
|AllCellsDistanceData.txt | |||
|top-level folder | |||
|Matrix of pairwise normalized Euclidean distances between cluster distribution vectors of all cells. | |||
|- | |||
|AllGroupsDistanceData.txt | |||
|top-level folder | |||
|Matrix of pairwise normalized Euclidean distances between average cluster distribution vectors of all groups. | |||
|- | |||
|AllGroupsSimilarityNetworkData.txt | |||
|top-level folder | |||
|Similarity network suitable for import and visualization in Cytoscape. | |||
|- | |||
|} | |||
<br><br> | |||
* '''<u>Configuration Parameters:</u>'''<br> | |||
{|class="wikitable" style="margin: auto;" | |||
|style="color:black; background-color:#ffffcc; width=15%"|'''Parameter Name''' | |||
|style="color:black; background-color:#ffffcc; width=15%"|'''Possible Values''' | |||
|style="color:black; background-color:#ffffcc; width=30%"|'''Description''' | |||
|- | |||
|''Image File Folder'' | |||
| | |||
|Top-level folder containing data for all groups and experimental conditions of interest, respectively. | |||
|- | |||
|rowspan=2|''Boundary File Format'' | |||
|LABEL_IMAGE | |||
|rowspan=2|Format of the segmentation data files with cell boundary information:<br> - LABEL_IMAGE: images with unique labels for each cell and a value of zero for the background <br> - IJ_ROIS: set of ImageJ 1.x ROIs, one ROI for each cell | |||
|- | |- | ||
| | |IJ_ROIS | ||
|- | |- | ||
|'' | |''Cytoskeleton Channel'' | ||
| | | | ||
|Channel with the image data of the fluorescently labeled cytoskeleton. | |||
|- | |- | ||
|'' | |''Calculate features'' | ||
| | | | ||
|Activates the feature calculation, can be omitted if features have been extracted already. | |||
|- | |- | ||
|'' | |''Feature Extractor'' | ||
| | | | ||
|Feature operator to apply. | |||
|- | |- | ||
|'' | |''Tile size'' | ||
| | | | ||
|Tile size in x and y direction for the sliding window used for feature extraction. | |||
|- | |- | ||
|'' | |''Tile shift'' | ||
| | | | ||
|Tile shifts in x and y direction, i.e. pixel distance between subsequent positions of the sliding window. | |||
|- | |- | ||
|''Number of feature clusters'' | |''Number of feature clusters'' | ||
| | | | ||
|Number of feature clusters applied in feature vector clustering. | |||
|- | |- | ||
|''Do PCA in stage II?'' | |''Do PCA in stage II?'' | ||
| | | | ||
|Optionally, a principal component analysis (PCA) can be applied to the extracted cluster distribution vectors,<br> and subsequent distance calculations can be restricted to the most significant principal components only. | |||
|} | |} | ||
===== Remarks and Important Notes ===== | |||
* The ''CytoskeletonAnalyzer2D'' extracts group names from the directory names. Thereby underscores are interpreted as separators. Thus, do not name your groups with common prefixes before the first underscore. For example, instead of naming your groups 'mygroup' and 'mygroup_variant', name them 'mygroup' and 'mygroupVariant' or something similar. To be completely on the save side, avoid underscores completely. | |||
<!-- | |||
===== Additional Tools ===== | ===== Additional Tools ===== | ||
The hierarchical clustering in stage II of our approach as described in the paper has been done using the [http://deim.urv.cat/~sgomez/multidendrograms.php MultiDendrograms] software.<br> In principal every hierarchical clustering tool can be applied. <br> | The hierarchical clustering in stage II of our approach as described in the paper has been done using the [http://deim.urv.cat/~sgomez/multidendrograms.php MultiDendrograms] software.<br> In principal every hierarchical clustering tool can be applied. <br> | ||
| Line 97: | Line 148: | ||
You can download the latest version of MultiDendrograms from its webpage: [http://deim.urv.cat/~sgomez/multidendrograms.php] | You can download the latest version of MultiDendrograms from its webpage: [http://deim.urv.cat/~sgomez/multidendrograms.php] | ||
--> | |||
<!-- | |||
===== Sample data ===== | ===== Sample data ===== | ||
For testing the ''ActinAnalyzer2D'' operator we provide some sample data: | For testing the ''ActinAnalyzer2D'' operator we provide some sample data: | ||
| Line 117: | Line 170: | ||
'''''IGF2BP1 promotes mesenchymal cell properties and migration of tumor-derived cells by enhancing the expression of LEF1 and SNAI2 (SLUG)'''''<br> | '''''IGF2BP1 promotes mesenchymal cell properties and migration of tumor-derived cells by enhancing the expression of LEF1 and SNAI2 (SLUG)'''''<br> | ||
''Nucleic Acids Res. Jul 2013; 41(13): 6618–6636. Published online May 15, 2013. doi: 10.1093/nar/gkt410'', [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3711427/ Article] | ''Nucleic Acids Res. Jul 2013; 41(13): 6618–6636. Published online May 15, 2013. doi: 10.1093/nar/gkt410'', [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3711427/ Article] | ||
--> | |||
=== Updates === | === Updates === | ||
----<br> | ----<br> | ||
''' | '''December 2020''' | ||
* Released first official version of Cytoskeleton Analyzer 2D. | * Released some improvements and small bug fixes with regard to handling ImageJ ROI files in MiToBo/MiToBo plugins 2.1. | ||
'''December 2018''' | |||
* Released first official version of Cytoskeleton Analyzer 2D in MiToBo/MiToBo plugins 1.8.13. | |||
<br> | |||
<br> | |||
== Visualization and Evaluation of Result Data in R == | |||
One option for further analyzing the outcomes of the Cytoskeleton Analyzer 2D, particularly differences and similarities between pattern distribution vectors, is to generate heatmaps of the pairwise distances between cell or group distribution vectors, respectively. | |||
As a starting point you can use the following R script: | |||
* [https://mitobo.informatik.uni-halle.de/downloads/cytoskeletonAnalyzer/generateDistanceHeatmaps.R generateDistanceHeatmaps.R] | |||
The script generates two heatmap plots, one to compare individual cells against each other and one to compare the average distribution vectors of different groups. | |||
===== Usage ===== | |||
* Download the script file to your local system. | |||
You can either use RStudio or R directly to execute the script. | |||
====== RStudio ====== | |||
* Run [https://www.rstudio.com/ RStudio] and open the script file in the editor window. | |||
* Edit lines 8 and 10 and enter the paths to your distance data files resulting from analyzing your data with the Cytoskeleton Analyzer 2D (see above). | |||
* Enter the destinations of the output files in lines 15 and 17 of the script. | |||
* To run the script, click the "Source" button in the top right corner of the RStudio GUI. | |||
====== R ====== | |||
If you prefer to use plain R you can specify the input and output data file names in the script using your favorite text editor, and then run the script directly in R. | |||
Latest revision as of 16:53, 1 December 2020
Cytoskeleton Analyzer 2D

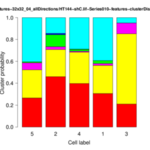
The Cytoskeleton Analyzer 2D is available since release version 1.8.13 of MiToBo.
This operator is an extended version of the Actin Analyzer 2D operator which was released in MiToBo version 1.4. The new version provides local binary patterns as new texture features and has received improvements with regard to user-friendliness. In addition, to ease the annotation of cell areas which is an essential prerequisite for applying the Cytoskeleton Analyzer, a supplemental plugin for cell contour segmentation and a handy interactive editor for label images have been released.
Latest News
The Cytoskeleton Analyzer Plugin has been released in MiToBo and MiToBo-Plugins 1.8.13.
Related Publications
- K. Bürstenbinder, B. Möller, R. Plötner, G. Stamm, G. Hause, D. Mitra, and S. Abel,
"The IQD Family of Calmodulin-Binding Proteins Links Calcium Signaling to Microtubules, Membrane Subdomains, and the Nucleus".
In Plant Physiology, 173(3):1692-1708, March 2017.
Name of Plugin/Operator
de.unihalle.informatik.MiToBo.apps.cytoskeleton.CytoskeletonAnalyzer2D
(available since MiToBo version 1.8.13)
Main features
- automatic extraction of different structural patterns by unsupervised texture analysis and clustering
- co-occurence matrices and Haralick features as well as local binary patterns are available for texture characterization
- structure quantification performed based on cell-wise cluster distributions
Usage
To run the CytoskeletonAnalyzer2D perform the following steps:
- install MiToBo by following the instructions on the Installation page
- run MiToBo and start the operator runner by selecting the menu item MiToBo Runner from Plugins -> MiToBo
- in the selection menu navigate to 'de.unihalle.informatik.MiToBo.apps.cytoskeleton' and select the operator CytoskeletonAnalyzer2D
This will bring up the operator window of the CytoskeletonAnalyzer2D.
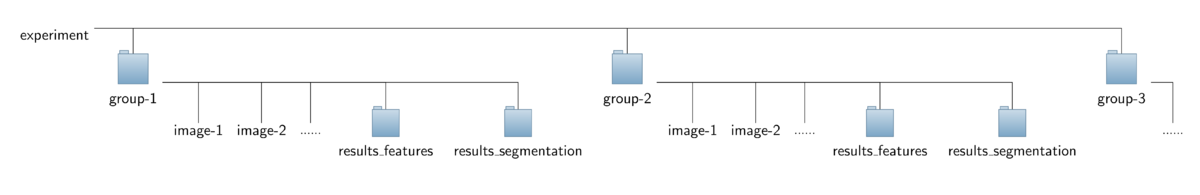
- Input data:
The operator expects a special organization of the input image data which is shown in the figure below. All the data should be contained in a common top-level folder, here named "experiment".
The images for each treatment/genotype/protein have to be put into separate sub-folders of this top-level folder, here named "group-1", "group-2" and so on. Besides the set of corresponding images,
each sub-folder is required to contain an additional sub-folder named "results_segmentation" where the annotation files with the cell areas or boundaries, respectively, are stored.
- For each image named "<imagename>.tif" a corresponding mask file is expected to be found in that folder. The mask file should have the same basename like the corresponding image file,
but either end on "-mask.tif" (e.g., "<imagename>-mask.tif") in case of using label images as masks, or on "-mask.zip" or "-mask.roi" (e.g., "<imagename>-mask.zip"), respectively, in case of using region sets as mask data.
Note that the operator currently only accepts ImageJ 1.x ROI sets as input. If label images are used as segmentation masks, the labels of individual cells in the label images are used as unique identifiers for the cells, i.e.,
are used as labels in the various plots. If the segmentation data is given in terms of ROIs a unique identifier is derived from the order of the cell boundaries in the ROI set.
Note that in the latter case a mask image is written to the output directory as additional output where the cells are marked by their identifiers to allow for easier assessment of the results.
Each of the sub-folders in the top-level folder is treated as a separate group of cells or experimental condition, thus, the cells of all images within one sub-folder are collected in a single set.
The names of the folders are used to label the different groups in the various output data files.
If the input images contain more than one channel you can select the channel on which the Cytoskeleton Analyzer should work. By default the first channel is used.
- Output data:
The operator generates image- or group-specific output data files as listed in the table below. All data files specific to a single input image are stored in a new sub-folder named "results_features" in the sub-folder of each group. All global output data files are stored directly in the top-level folder. The string imageID below represents the name of a single input image, the string groupID refers to a specific group. In addition to saving the output data to file bar charts and box-whisker plots are directly shown in the graphical user interface of ImageJ/Fiji upon termination of the analysis process.
| Output File Name | Where to find it... | Description |
| <imageID>-features.txt | folder results_features | Feature data for single image file. |
| <imageID>-features.tif | folder results_features | Image stack visualizing the feature data for single image. |
| <imageID>-clusterDistro.txt | folder results_features | Cluster distributions for each cell individually and for all cells in total of the image. |
| <imageID>-clusters.tif | folder results_features | Pseudo-colored image illustrating the cluster distribution per image. |
| <groupID>-distributionChart.png | folder results_features | Stacked bar plot of the cluster distribution for each cell of the group. |
| AllCellsClusterStats.txt | top-level folder | Cluster distribution of raw data for all images and cells. |
| AllCellsPCASubspaceStats.txt | top-level folder | If PCA is applied to the cluster distribution vectors this file contains the subspace feature vectors for all cells. |
| AllCellsDistanceData.txt | top-level folder | Matrix of pairwise normalized Euclidean distances between cluster distribution vectors of all cells. |
| AllGroupsDistanceData.txt | top-level folder | Matrix of pairwise normalized Euclidean distances between average cluster distribution vectors of all groups. |
| AllGroupsSimilarityNetworkData.txt | top-level folder | Similarity network suitable for import and visualization in Cytoscape. |
- Configuration Parameters:
| Parameter Name | Possible Values | Description |
| Image File Folder | Top-level folder containing data for all groups and experimental conditions of interest, respectively. | |
| Boundary File Format | LABEL_IMAGE | Format of the segmentation data files with cell boundary information: - LABEL_IMAGE: images with unique labels for each cell and a value of zero for the background - IJ_ROIS: set of ImageJ 1.x ROIs, one ROI for each cell |
| IJ_ROIS | ||
| Cytoskeleton Channel | Channel with the image data of the fluorescently labeled cytoskeleton. | |
| Calculate features | Activates the feature calculation, can be omitted if features have been extracted already. | |
| Feature Extractor | Feature operator to apply. | |
| Tile size | Tile size in x and y direction for the sliding window used for feature extraction. | |
| Tile shift | Tile shifts in x and y direction, i.e. pixel distance between subsequent positions of the sliding window. | |
| Number of feature clusters | Number of feature clusters applied in feature vector clustering. | |
| Do PCA in stage II? | Optionally, a principal component analysis (PCA) can be applied to the extracted cluster distribution vectors, and subsequent distance calculations can be restricted to the most significant principal components only. |
Remarks and Important Notes
- The CytoskeletonAnalyzer2D extracts group names from the directory names. Thereby underscores are interpreted as separators. Thus, do not name your groups with common prefixes before the first underscore. For example, instead of naming your groups 'mygroup' and 'mygroup_variant', name them 'mygroup' and 'mygroupVariant' or something similar. To be completely on the save side, avoid underscores completely.
Updates
December 2020
- Released some improvements and small bug fixes with regard to handling ImageJ ROI files in MiToBo/MiToBo plugins 2.1.
December 2018
- Released first official version of Cytoskeleton Analyzer 2D in MiToBo/MiToBo plugins 1.8.13.
Visualization and Evaluation of Result Data in R
One option for further analyzing the outcomes of the Cytoskeleton Analyzer 2D, particularly differences and similarities between pattern distribution vectors, is to generate heatmaps of the pairwise distances between cell or group distribution vectors, respectively.
As a starting point you can use the following R script:
The script generates two heatmap plots, one to compare individual cells against each other and one to compare the average distribution vectors of different groups.
Usage
- Download the script file to your local system.
You can either use RStudio or R directly to execute the script.
RStudio
- Run RStudio and open the script file in the editor window.
- Edit lines 8 and 10 and enter the paths to your distance data files resulting from analyzing your data with the Cytoskeleton Analyzer 2D (see above).
- Enter the destinations of the output files in lines 15 and 17 of the script.
- To run the script, click the "Source" button in the top right corner of the RStudio GUI.
R
If you prefer to use plain R you can specify the input and output data file names in the script using your favorite text editor, and then run the script directly in R.