Applications/CytoskeletonAnalyzer2D
Cytoskeleton Analyzer 2D

The Cytoskeleton Analyzer 2D is available since release version 1.8.13 of MiToBo.
This operator is an extended version of the Actin Analyzer 2D operator which was released in MiToBo version 1.4. The new version provides local binary patterns as new texture features and has received improvements with regard to user-friendliness. In addition, to ease the annotation of cell areas which is an essential prerequisite for applying the Cytoskeleton Analyzer, a supplemental plugin for cell contour segmentation and a handy interactive editor for label images have been released.
Latest News
The Cytoskeleton Analyzer Plugin has been released in MiToBo and MiToBo-Plugins 1.8.13.
Related Publications
- K. Bürstenbinder, B. Möller, R. Plötner, G. Stamm, G. Hause, D. Mitra, and S. Abel,
"The IQD Family of Calmodulin-Binding Proteins Links Calcium Signaling to Microtubules, Membrane Subdomains, and the Nucleus".
In Plant Physiology, 173(3):1692-1708, March 2017.
Name of Plugin/Operator
de.unihalle.informatik.MiToBo.apps.cytoskeleton.CytoskeletonAnalyzer2D
(available since MiToBo version 1.8.13)
Main features
- automatic extraction of different structural patterns by unsupervised texture analysis and clustering
- co-occurence matrices and Haralick features as well as local binary patterns are available for texture characterization
- structure quantification performed based on cell-wise cluster distributions
Usage
To run the CytoskeletonAnalyzer2D perform the following steps:
- install MiToBo by following the instructions on the Installation page
- run MiToBo and start the operator runner by selecting the menu item MiToBo Runner from Plugins -> MiToBo
- in the selection menu navigate to 'de.unihalle.informatik.MiToBo.apps.cytoskeleton' and select the operator CytoskeletonAnalyzer2D
This will bring up the operator window of the CytoskeletonAnalyzer2D.
- Input data:
The operator expects a special organization of the input image data. All the data should be contained in common top-level folder.
The images for each treatment/genotype/protein have to be put into a separate sub-folder of this top-level folder. Besides the set of corresponding images,
each sub-folder is required to contain an additional sub-folder named "results_segmentation" where the annotation files with the cell areas or boundaries, respectively, are stored.
For each image named "<imagename>.tif" a corresponding mask file is expected to be found in that folder. The mask file should have the same basename like the corresponding image file,
but either end on "-mask.tif" (e.g., "<imagename>-mask.tif") in case of using label images as masks, or on "-mask.zip" or "-mask.roi" (e.g., "<imagename>-mask.zip"), respectively, in case of using region sets as mask data.
Note that the operator currently only accepts ImageJ 1.x ROI sets as input. If label images are used as segmentation masks, the labels of individual cells in the label images are used as unique identifiers for the cells, i.e.,
are used as labels in the various plots. If the segmentation data is given in terms of ROIs a unique identifier is derived from the order of the cell boundaries in the ROI set.
Note that in the latter case a mask image is written to the output directory as additional output where the cells are marked by their identifiers to allow for easier assessment of the results.
Each of the sub-folders in the top-level folder is treated as a separate group of cells or experimental conditions, thus, the cells of all images within one folder a collected in a single set. The names of the folders are used to label the different groups in the various output data files.
If the input images contain more than one channel you can select the channel on which the Cytoskeleton Analyzer should work. By default the first channel is used.
- Output data:
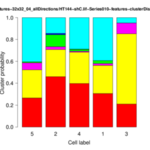
The operator displays the cluster distributions for each group of cells as stacked bar plots and box-whisker plots. In addition, it writes several files to the given output folder:- *-features.txt: feature data for each image
- *-features.tif: image stack visualizing the feature data
- *-features-config.ald: configuration of the operator in this run
- *-clusterDistro.txt: cluster distributions per image
- *-clusters.tif: pseudo-colored image illustrating the cluster distribution per image
- AllImagesClusterStatistics.txt: cluster distribution raw data for all images
- AllImagesSubspaceFeatures.txt: if PCA is applied to the cluster distributions prior to the distance calculations, this file contains the subspace feature vectors
- AllImagesPairwiseDistanceData.txt: matrix of pairwise Euclidean distances for distribution vectors, can be examined, e.g., with Multidendrograms (see below)
- *-distribution.png: for each cell group a stacked bar plot is saved showing the cluster distribution for each cell of the group
- Parameters:
| Name | Description |
| Image directory | directory where the input image data can be found |
| Mask directory | directory where the label images or region boundary files can be found |
| Mask format | format of the segmentation data files: LABEL_IMAGE = images with unique labels for each cell and a value of zero for the background / IJ_ROIS = set of ImageJ 1.x ROIs, one ROI for each cell |
| Output and working directory | directory to which the result files and intermediate data is written |
| Calculate features | if disabled the operator expects the features to be already present in the input directory and skips the (time-consuming) feature calculations; this option is helpful if the features have already been calculated ones and only the parameters of the clustering should be changed |
| Feature directory | directory where the features should be saved or - in case they are already available - from where they are read; the directory can be the same as the output and working directory |
| Tile size x/y | size of the sliding window used for feature calculations, should be chosen according to the resolution of the input images |
| Tile shift x/y | shift of the sliding window, if the shift is smaller than the tile size sliding windows overlap |
| Distance | pixel-pair distance in co-occurence matrix calculations |
| Set of directions | directions to be considered in co-occurence matrix calculations |
| Isotropic calculations | the texture features are derived from co-occurence matrixes; if this flag is enabled features for different directions are averaged, otherwise all individual directions are preserved (resulting in larger, but also more informative feature vectors) |
| Number of feature clusters | number of clusters in first stage, i.e., number of expected structural patterns in the images |
| Do PCA in stage II? | allows to enable/disable PCA on the cluster distribution vectors prior to the pairwise distance calculations; by default enabled |
Additional Tools
The hierarchical clustering in stage II of our approach as described in the paper has been done using the MultiDendrograms software.
In principal every hierarchical clustering tool can be applied.
The basis for the hierarchical cluster analysis is the file AllImagesPairwiseDistanceData.txt to be found in the output directory upon termination of an analysis run. It contains a matrix of pairwise Euclidean distances of the (optionally dimension-reduced) cluster distribution vectors of all cells. The file can directly be loaded by MultiDendrograms, for other tools format convertion might be necessary.
You can download the latest version of MultiDendrograms from its webpage: [1]
Sample data
For testing the ActinAnalyzer2D operator we provide some sample data:
(With release 1.8 the handling of file names and automatic deduction of mask file names changed, thus, image names in the sample data archive had to be updated.)
The archive contains the following sub-folders:
- imageData: test images that were used in the ICPR publication mentioned above
- maskData: corresponding label images
- featureData: pre-calculated features for the cells (re-calculating the features may require up to an hour, depending on the machine used)
- resultData: sample results calculated on the given data
In addition, in the archive a file with a sample parameter configuration for the operator can be found. The parameters are those used for producing the sample results. Once the operator has been started the file can be loaded via the 'File' menu and its entry 'Load Settings' . Note that you need to set the image and mask directories, and also the feature directory according to your local file system structure and the place to where you extracted the zip file.
For more information on the data and the morphological analysis of the cells, see
Anne Zirkel, Marcell Lederer, Nadine Stöhr, Nikolaos Pazaitis, and Stefan Hüttelmaier
IGF2BP1 promotes mesenchymal cell properties and migration of tumor-derived cells by enhancing the expression of LEF1 and SNAI2 (SLUG)
Nucleic Acids Res. Jul 2013; 41(13): 6618–6636. Published online May 15, 2013. doi: 10.1093/nar/gkt410, Article
Updates
November 2018
- Released first official version of Cytoskeleton Analyzer 2D.